Quotidiano on line
di informazione sanitaria
Giovedì 25 APRILE 2024
Scienza e Farmacidi informazione sanitaria
Giovedì 25 APRILE 2024
Cosa dice la nuova guida dell'Ema sui biosimilari
07 MAG - L’Ue è pioniera dei biosimiliari: ha iniziato nel 2006 ed è stata la prima a pensare alle misure della loro regolazione.
L’Ema ha realizzato la prima guida su questo tipo di farmaci in considerazione che negli ultimi 10 anni l'Ue ha approvato il maggior numero di biosimilari in tutto il mondo, sommando notevoli esperienze di utilizzo e sicurezza che dimostrano che i biosimilari approvati secondo le procedure EMA possono essere utilizzati in modo sicuro ed efficace in tutte le loro indicazioni approvate per altri farmaci biologici.
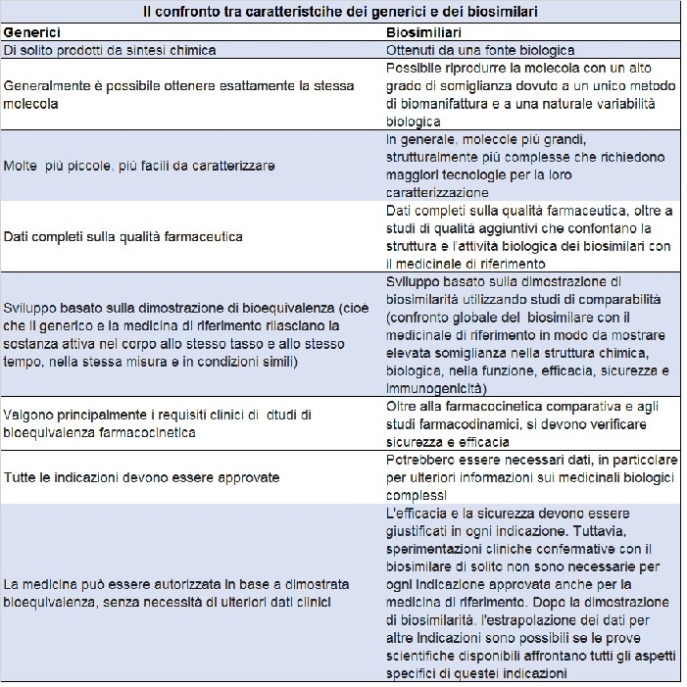
Un biosimilare, spiega la guida, è una medicina biologica molto simile a un'altra medicina biologica già approvata nell'Unione europea (la cosiddetta medicina di riferimento). Poiché però i biosimilari sono basati su materiali biologici, potrebbero esserci alcune differenze minori rispetto alla medicina di riferimento. Queste però non sono clinicamente significative, e non cambiano affatto rispetto all’originale in termini di sicurezza e efficacia.
La variabilità naturale riguarda tutti i medicinali biologici e controlli rigorosi sono sempre in atto per garantire che questa non influenzi gli effetti o la sicurezza.
I biosimilari sono approvati secondo gli stessi standard di qualità farmaceutica, sicurezza ed efficacia di tutti i medicinali biologici approvati nell'Ue.
Dimostrando la biosimilarità, un farmaco può contare sulle stesse prove di sicurezza e di efficacia della medicina di riferimento. Qusto evita la ripetizione inutile di prove cliniche già eseguite.
La dimostrazione della biosimilarità si basa su studi di comparabilità completi con il medicinale di riferimento. Se un biosimilare è molto simile a un medicinale di riferimento e ha una sicurezza e un'efficacia paragonabili per una indicazione terapeutica, si possono utilizzare tutte le procedure di controllo già utilizzate per il medicinale di riferimento.
Questo non è un concetto nuovo, ma un principio scientifico ben consolidato utilizzato di routine quando i medicinali biologici con diverse indicazioni approvate subiscono importanti modifiche alla loro produzione (ad esempio per introdurre una nuova formulazione). Nella maggior parte di questi casi, le sperimentazioni cliniche non vengono ripetute e tutte le indicazioni e le modifiche sono approvate sulla base di studi di qualità e di confronto in vitro. Tutte le indicazioni dei medicinali biologici (compresi i biosimilari) sono state determinate in base a prove scientifiche.
La sicurezza dei biosimilari è monitorata attraverso attività di farmacovigilanza, esattamente come per le altre medicine. Non esiste alcuna specifica esigenza di sicurezza applicabile solo ai iosimilari legata al loro percorso di sviluppo diverso.
L'Ema non regola l'intercambiabilità e la sostituzione di un medicinale di riferimento col suo biosimilare Queste rientrano nel campo di competenza degli Stati membri.
La Guida Ema descrive poi le caratteristiche specifiche dei medicinali biosimilari.
Sono anzitutto molto simili alla medicina di riferimento. Il biosimilare ha proprietà fisiche, chimiche e biologiche molto simili al medicinale di riferimento. Ci possono essere differenze minori che non sono clinicamente significative in termini di sicurezza o efficacia.
Non c’è alcun effetto clinico differente rispetto alla medicina di riferimento. Non ci sono differenze nell’efficacia clinica. Gli studi clinici per sostenere l'approvazione di un biosimilare confermano che le differenze non incidono sulla sicurezza e sull'efficacia.
La variabilità dei biosimilari va mantenuta entro limiti rigorosi. La minore variabilità è consentita solo quando le prove scientifiche dimostrano che non influenza la sicurezza e l'efficacia. La gamma di variabilità consentita per un biosimilare è la stessa di quella consentita tra i lotti di riferimento dei medicinali.
Stessi standard rigorosi di qualità, di sicurezza ed efficacia. I biosimilari sono approvati secondo gli stessi standard rigorosi di qualità, sicurezza e efficacia che si applicano a qualsiasi altro medicinale.
La guida spiega anche come i professionisti sanitari possono contribuire a migliorare la farmacovigilanza per i medicinali biologici.
È importante che il marchio della medicina e Il numero di lotto siano conosciuti da professionisti sanitari per l’utilizzo a tutti i livelli, inclusa l'erogazione e la somministrazione al paziente. I prescrittori dovrebbero includere il nome commerciale del farmaco nella ricetta. I professionisti della sanità devono garantire che il nome commerciale e il numero di lotto siano riportati In caso di reazioni negative sospette, secondo i regolamenti nazionali. Nei casi in cui il prodotto è dispensato da una farmacia, il nome del marchio e il numero di lotto del biologico dovrebbe essere fornito al paziente con la medicina. Se viene cambiata a un paziente una medicina biologica con un'altra con lo stesso principio attivo, è importante registrare il nome, il marchio e il numero di lotto per ciascuno dei farmaci. I professionisti della sanità dovrebbero contattare le autorità regolatorie del proprio Paese per consigli su come segnalare le reazioni avverse dei famaci.
Un capitolo della guida riguarda la comunicazione ai pazienti sui biosimilari.
Se i pazienti hanno domande se una medicina biologica sia un biosimilare, professionisti possono trovare queste informazioni nella SMPC, Summary Of Product Characteristics, della Commissione europea. Il foglietto illustrativo, che contiene raccomandazioni per i pazienti su come utilizzare la medicina correttamente, non include la menzione di biosimilarità, in quanto questa si riferisce solo allo sviluppo della medicina e non è correlato al suo utilizzo.
Se i pazienti che ricevono biosimilari in un ambiente clinico (ad esempio in ospedale) vogliono informazioni sul loro farmaco, possono chiedere ai professionisti che li seguono.
Il foglietto illustrativo può anche essere scaricato dal sito web di Ema.
Per le domande dei pazienti su cosa sono i biosimilari e come siano garantiti la sicurezza e l'efficacia, questi possono consultare le domande e risposte nella loro lingua, disponibili per il territorio europeo sul sito web dell Commissione Ue.
Quando un nuovo medicinale è approvato da Ema, l'Agenzia pubblica un riepilogo per il pubblico in cui spiega perché è stato approvato il farmaco. Queste documentazioni di sintesi (chiamate "sintesi EPAR"), sono disponibili sulla pagina di ciascun farmaco sul sito web di Ema sotto forma di questionario in tutte le lingue ufficiali dell'Ue.
È possibile accedere ai riepiloghi EPAR per i biosimilari cercando il nome della medicina sull’homepage di EMA.
Anche le autorità nazionali di regolamentazione forniscono Informazioni sui biosimilari nella loro lingua locale.
07 maggio 2017
© Riproduzione riservata
gli speciali

Quotidianosanità.it
Quotidiano online
d'informazione sanitaria.
QS Edizioni srl
P.I. 12298601001
Sede legale:
Via Giacomo Peroni, 400
00131 - Roma
Sede operativa:
Via della Stelletta, 23
00186 - Roma
Quotidiano online
d'informazione sanitaria.
QS Edizioni srl
P.I. 12298601001
Sede legale:
Via Giacomo Peroni, 400
00131 - Roma
Sede operativa:
Via della Stelletta, 23
00186 - Roma
Direttore responsabile
Luciano Fassari
Direttore editoriale
Francesco Maria Avitto
Luciano Fassari
Direttore editoriale
Francesco Maria Avitto
Tel. (+39) 06.89.27.28.41
info@qsedizioni.it
redazione@qsedizioni.it
Coordinamento Pubblicità
commerciale@qsedizioni.it
info@qsedizioni.it
redazione@qsedizioni.it
Coordinamento Pubblicità
commerciale@qsedizioni.it
- Joint Venture
- SICS srl
- Edizioni
Health Communication srl
Copyright 2013 © QS Edizioni srl. Tutti i diritti sono riservati
- P.I. 12298601001
- iscrizione al ROC n. 23387
- iscrizione Tribunale di Roma n. 115/3013 del 22/05/2013
Riproduzione riservata.
Policy privacy
- P.I. 12298601001
- iscrizione al ROC n. 23387
- iscrizione Tribunale di Roma n. 115/3013 del 22/05/2013
Riproduzione riservata.
Policy privacy