
Lo studio della dinamica molecolare non è mai stato così veloce
15 Maggio 2013
© Riproduzione riservata

Ebola, hantavirus, caldo estremo: l’Oms lancia l’allarme su tre fronti. “L’epidemia in Congo corre più veloce della risposta”
L'Organizzazione Mondiale della Sanità ha tracciato oggi il bilancio di tre emergenze sanitarie che tengono banco in diverse parti del mondo. Nel briefing stampa di questa mattina, il direttore generale...

Caldo e diabete, il caso Zverev ricorda i rischi dell’estate: “Attenzione ai sensori per la glicemia”
Una lettura della glicemia completamente sballata, una dose di insulina superiore al necessario e una grave ipoglicemia da correggere durante la partita con l'equivalente di dieci lattine di cola. L'episodio...

Ebola. Primo caso importato in Europa. L’Ecdc: “Il rischio di trasmissione sostenuta è molto basso, ma serve preparazione”
Il primo caso importato di Ebola in Europa dall'inizio dell'attuale epidemia è stato confermato in Francia. Un medico, rientrato dalla Repubblica Democratica del Congo, è risultato positivo al virus. La...

Hantavirus. Gli Usa chiudono il monitoraggio: nessun caso positivo. Kennedy Jr.: “Proteggere gli americani è la nostra priorità assoluta”
Il Dipartimento della Salute e dei Servizi Umani degli Stati Uniti (Hhs) ha annunciato oggi la conclusione della risposta sanitaria federale all'esposizione al virus Hantavirus associata alla nave da crociera...
Gli speciali

Sanità digitale per garantire più salute e sostenibilità. Ma servono standard e condivisione
I più letti
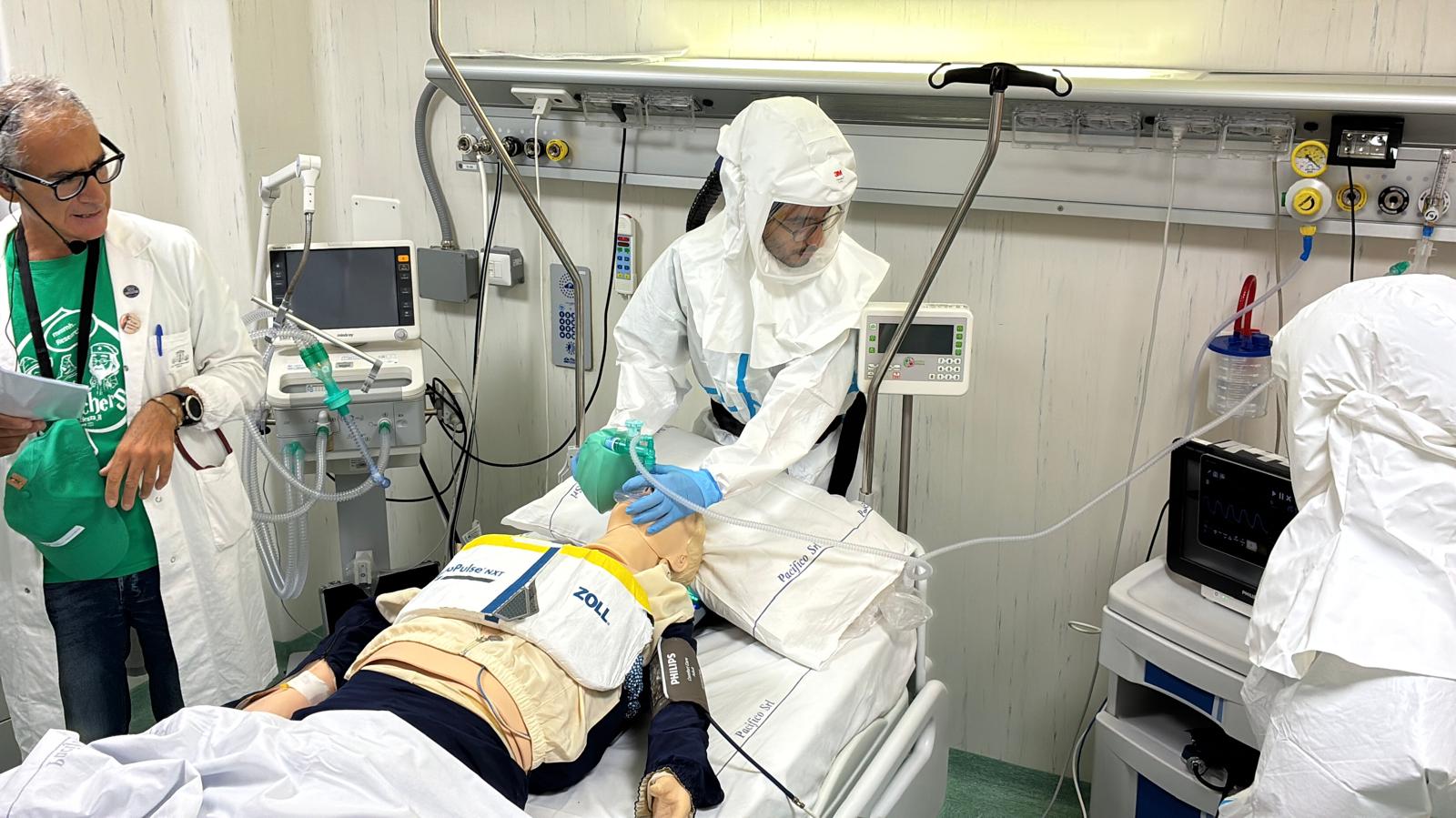
Emergenze infettive, alto isolamento e biocontenimento: Spallanzani e Aeronautica Militare insieme per formare gli operatori sanitari

Malattie venose, Servier lancia Daflon 1000 mg: una sola compressa per semplificare la terapia

Anziani non autosufficienti. Pronto il Piano nazionale 2025-2027: focus su domiciliarità, Pua, Leps e presa in carico integrata

“Il Ssn è governato come una fabbrica di prestazioni. E ogni giorno 10 professionisti lo abbandonano prima della pensione”. L'Anaao Assomed lancia tre proposte per riformare la sanità: “È ora di cambiare modello o crollerà”

Case di Comunità. Per i medici di famiglia un compenso di 38,72 euro l’ora con turni di almeno 3 ore continuative. Arriva la firma sul contratto tra la Sisac e i sindacati Fimmg e Fmt. No di Smi e Snami
Emergenze infettive, alto isolamento e biocontenimento: Spallanzani e Aeronautica Militare insieme per formare gli operatori sanitari
Malattie venose, Servier lancia Daflon 1000 mg: una sola compressa per semplificare la terapia

Rette RSA e Alzheimer: la giurisprudenza svolta. Per la prima volta un Tribunale condanna direttamente anche una Regione

Medico di famiglia. Anatomia di un fallimento

Enpam. Scontro M5S-Giorgetti sulle pensioni dei medici. Castellone: “Investimenti a rischio, c'è un'inchiesta della procura di Milano”