
Farmaci Biosimilari. Aifa aggiorna il Position Paper: “Intercambiabili con gli originatori, ma lo switch sia informato e tracciabile”
Nel terzo Position Paper l’Agenzia ribadisce il ruolo dei biosimilari per l’accesso alle cure e la sostenibilità del Ssn. Confermata la non sostituibilità automatica in farmacia, ma raccomandato lo switch informato con assenso del medico e adeguata informazione del paziente. IL DOCUMENTO
30 Aprile 2026
© Riproduzione riservata

Assemblea Farmindustria. Cattani: “Settore farmaceutico strategico per salute e crescita”. E poi al Governo: “Fissare limite al payback”. Bocciata la revisione del prontuario proposta da Aifa
Un settore industriale che corre più della media europea, sostiene l’export nazionale, investe in ricerca e produzione e rivendica un ruolo sempre più strategico non solo per la salute, ma...

Sangue, tessuti e cellule. L’Ecdc definisce linee guida unificate per lo screening delle epatiti B e C in Europa
Sangue, tessuti, cellule staminali e materiale riproduttivo più sicuri grazie a standard europei uniformi per lo screening delle epatiti B e C. Il Centro europeo per la prevenzione e il...

Prontuario farmaceutico. Nisticò: “La revisione serve alla sostenibilità del sistema. Più cure significano decisioni più complesse”
La revisione del Prontuario Farmaceutico Nazionale rappresenta un passaggio necessario per garantire la sostenibilità del Servizio sanitario nazionale in un contesto caratterizzato da una crescente complessità clinica e scientifica. È...

Oms: “L’ondata di calore in Europa è un’emergenza sanitaria”. Oltre 200mila morti in quattro anni
Non è solo una questione meteorologica. L'ondata di calore che da una settimana sta investendo diversi paesi europei, con temperature che in molte città potrebbero toccare i 40 gradi, è...
Gli speciali

Sanità digitale per garantire più salute e sostenibilità. Ma servono standard e condivisione
I più letti
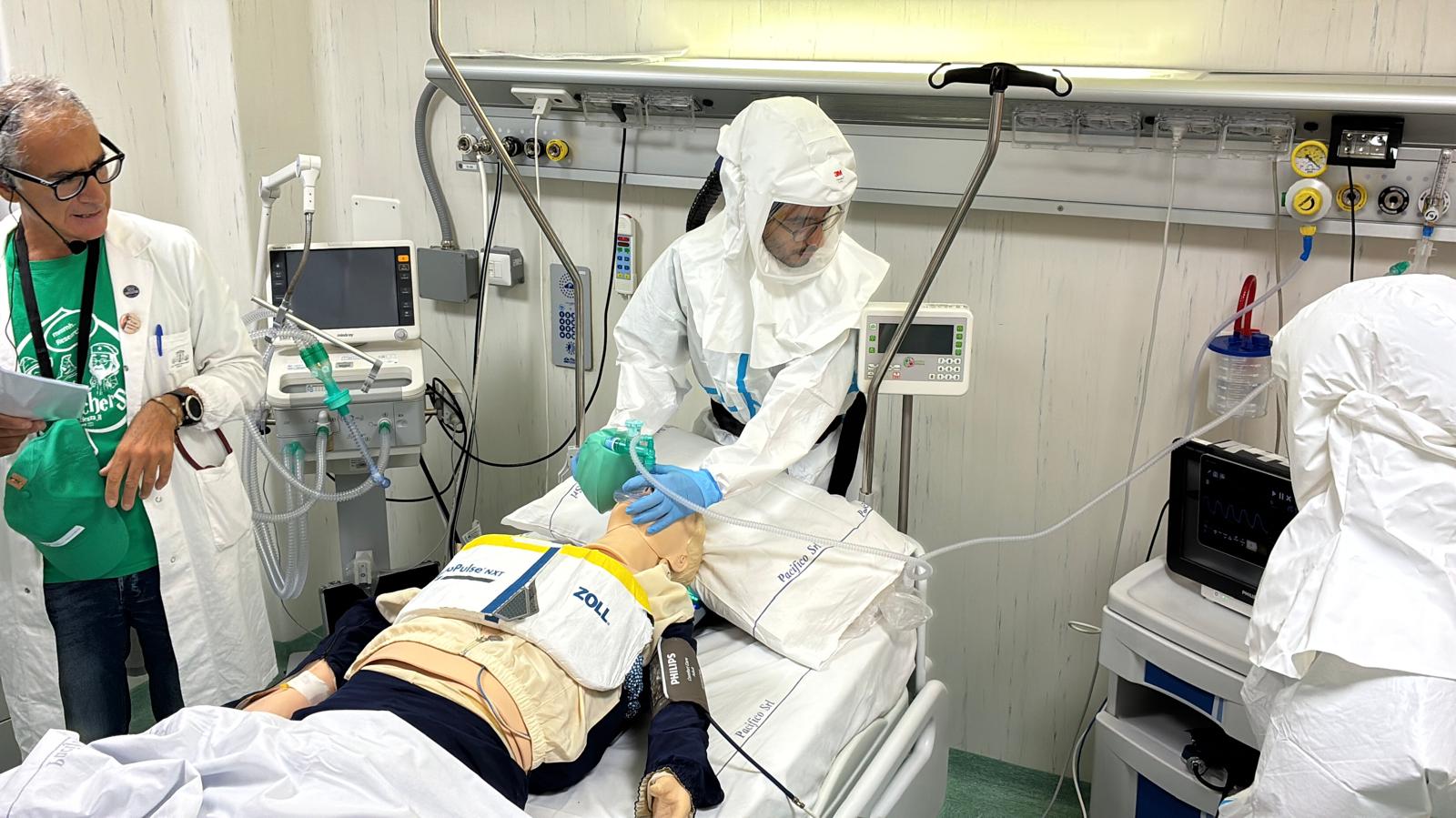
Emergenze infettive, alto isolamento e biocontenimento: Spallanzani e Aeronautica Militare insieme per formare gli operatori sanitari

“Il Ssn è governato come una fabbrica di prestazioni. E ogni giorno 10 professionisti lo abbandonano prima della pensione”. L'Anaao Assomed lancia tre proposte per riformare la sanità: “È ora di cambiare modello o crollerà”

Anziani non autosufficienti. Pronto il Piano nazionale 2025-2027: focus su domiciliarità, Pua, Leps e presa in carico integrata

Un accordo ad hoc per riempire le Case di Comunità e rispettare la scadenza del Pnrr. Obbligo per i medici di famiglia fino a 6 ore a settimana. Ecco la proposta di Regioni e Ministero della Salute

Medici di famiglia nelle Case della comunità. Previsto un compenso di circa 40 euro l’ora
Emergenze infettive, alto isolamento e biocontenimento: Spallanzani e Aeronautica Militare insieme per formare gli operatori sanitari

Rette RSA e Alzheimer: la giurisprudenza svolta. Per la prima volta un Tribunale condanna direttamente anche una Regione

Medico di famiglia. Anatomia di un fallimento

Enpam. Scontro M5S-Giorgetti sulle pensioni dei medici. Castellone: “Investimenti a rischio, c'è un'inchiesta della procura di Milano”

Dottori anche con la laurea triennale: perché continuare a sminuirli?