
Farmaci. Ecco le regole europee per i trial per uso umano condotti fuori UE
17 Aprile 2012
© Riproduzione riservata

Ema. Via libera a 12 nuovi farmaci, tra cui impianto oculare rivoluzionario e primo orale per la psoriasi
Il Comitato per i medicinali per uso umano (Chmp) dell'Agenzia europea per i medicinali (Ema), riunito dal 20 al 23 luglio 2026, ha raccomandato l'approvazione di dodici nuovi farmaci e...

Prontuario farmaceutico, il Governo verso il cambio di rotta. Stop alla revisione per classi terapeutiche: si punta a una “pulizia” mirata e a rinegoziazione dei prezzi
Una significativa correzione di rotta sulla revisione del Prontuario farmaceutico nazionale. È quanto sarebbe emerso, secondo quanto apprende Quotidiano Sanità, nel corso di un incontro che si è tenuto nei...

Disforia di genere, SIP e ACP rispondono al Garante Infanzia sulla guida contestata: “Strumento informativo e formativo, non una linea guida clinico-terapeutica”
La Società Italiana di Pediatria e l’Associazione Culturale Pediatri intervengono nel dibattito sviluppatosi intorno alla guida “Oltre lo sguardo. Guida pratica su varianza di genere, orientamenti sessuali e omogenitorialità per...
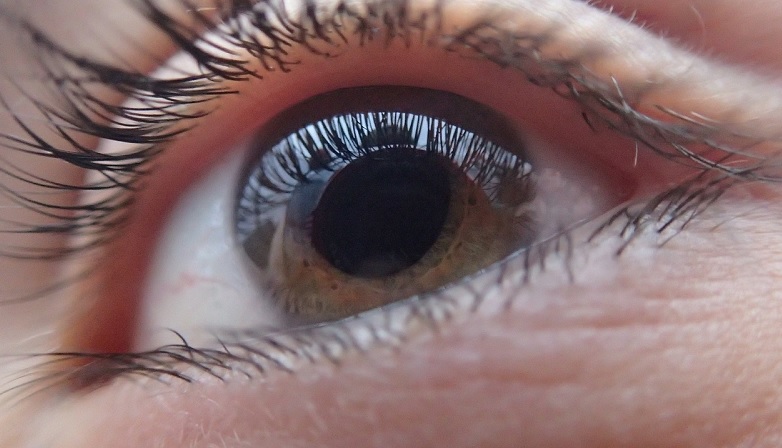
Cieca, torna a vedere grazie ad un trapianto di una parte della retina e del segmento anteriore da un occhio all’altro. Prima volta al mondo, presso le Molinette di Torino
Primato mondiale per la sanità italiana presso la Città della Salute e della Scienza di Torino. Per la prima volta nella storia della medicina, l'équipe della Clinica Oculistica universitaria dell'ospedale...
Ultime analisi e review da QS Pro Gold







I più letti

Aspiranti medici di famiglia, entrate al corso di formazione specifica con gli occhi aperti e piena consapevolezza

Enpam. Durigon: "Controlli in corso". M5S: "Compensi da 1 milione ai vertici e conflitti di interessi"

Lorenzin (Pd) a Schillaci: “La sanatoria no vax apre la strada all’interferenza della politica sulla deontologia medica”

Medici di famiglia: se un lavoro non lo sceglie quasi nessuno, il problema è il modello, non i medici

Estati torride, l'aria condizionata non è sostenibile sul lungo periodo. Gli scienziati cercano alternative

Benzodiazepine. Stop alla ricetta elettronica: funzionalità sospesa dopo il caos organizzativo

Riflessione di un Tecnico sanitario di radiologia medica
Lorenzin (Pd) a Schillaci: “La sanatoria no vax apre la strada all’interferenza della politica sulla deontologia medica”

Lombardia. Le immagini diagnostiche sul Fascicolo Sanitario Elettronico

Influenza. Via libera alla Circolare 2026-2027: vaccino raccomandato a partire dai 6 mesi. Soglia per i vaccini potenziati abbassata a 60 anni