
Malattie rare. Scoperta a firma italiana possibile cura per forme gravi di alfa-sarcoglicanopatia
È il primo e più ampio studio multicentrico al mondo, svolto su 16 pazienti, a concentrarsi su questa rara forma di distrofia muscolare caratterizzata da insorgenza precoce, in età infantile, e rapida progressione. Lo studio pubblicato su Brain ha permesso inoltre di identificare i pazienti che meglio potrebbero rispondere ai trattamenti
13 Gennaio 2026
© Riproduzione riservata

Caldo estremo, FADOI: “Sopra i 40 gradi il corpo può non riuscire più a disperdere il calore”
Le temperature estreme che stanno interessando gran parte dell’Italia non rappresentano soltanto un problema di comfort, ma una vera emergenza sanitaria. A lanciare l’allarme è FADOI, la Federazione delle Associazioni...

Ondate di calore, i consigli della SIP per proteggere i bambini
Le ondate di calore che stanno interessando il Paese possono rappresentare un rischio per la salute dei bambini, soprattutto nei primi anni di vita. I più piccoli hanno infatti una...

Demenza, da OMS nuove linee guida per ridurre il rischio: attenzione all’inquinamento
Oltre 57 milioni di persone al mondo convivono con la demenza, con 10 milioni di nuove diagnosi che si aggiungono ogni anno; di queste, l’Alzheimer rappresenta il 60-70% dei casi....

Roche lancia AXELIOS 1, nuova piattaforma di sequenziamento per espansione dedicata alla ricerca
Il sequenziamento di nuova generazione (NGS) ha rivoluzionato negli ultimi anni la ricerca biomedica, consentendo di ottenere informazioni sempre più dettagliate sul patrimonio genetico e sui meccanismi molecolari alla base...
Gli speciali

Sanità digitale per garantire più salute e sostenibilità. Ma servono standard e condivisione
I più letti

Riflessione di un Tecnico sanitario di radiologia medica

Lombardia. Le immagini diagnostiche sul Fascicolo Sanitario Elettronico

Cancro. Oms-Iarc: una persona su 5 svilupperà la malattia. Entro il 2050 attesi 35 mln di nuovi casi l’anno. Quasi il 40% è prevenibile, ma il vero divario è tra ciò che sappiamo e ciò che facciamo

Lea 2024: il Nord guida e il Sud insegue. Veneto, Emilia-Romagna e Toscana si confermano al top. Calabria, Sicilia e Bolzano in fondo alla classifica

Ebola. Secondo operatore umanitario statunitense contagiato, trasferito in Germania. Tedros: “Proteggere chi è in prima linea”

Benzodiazepine. Stop alla ricetta elettronica: funzionalità sospesa dopo il caos organizzativo
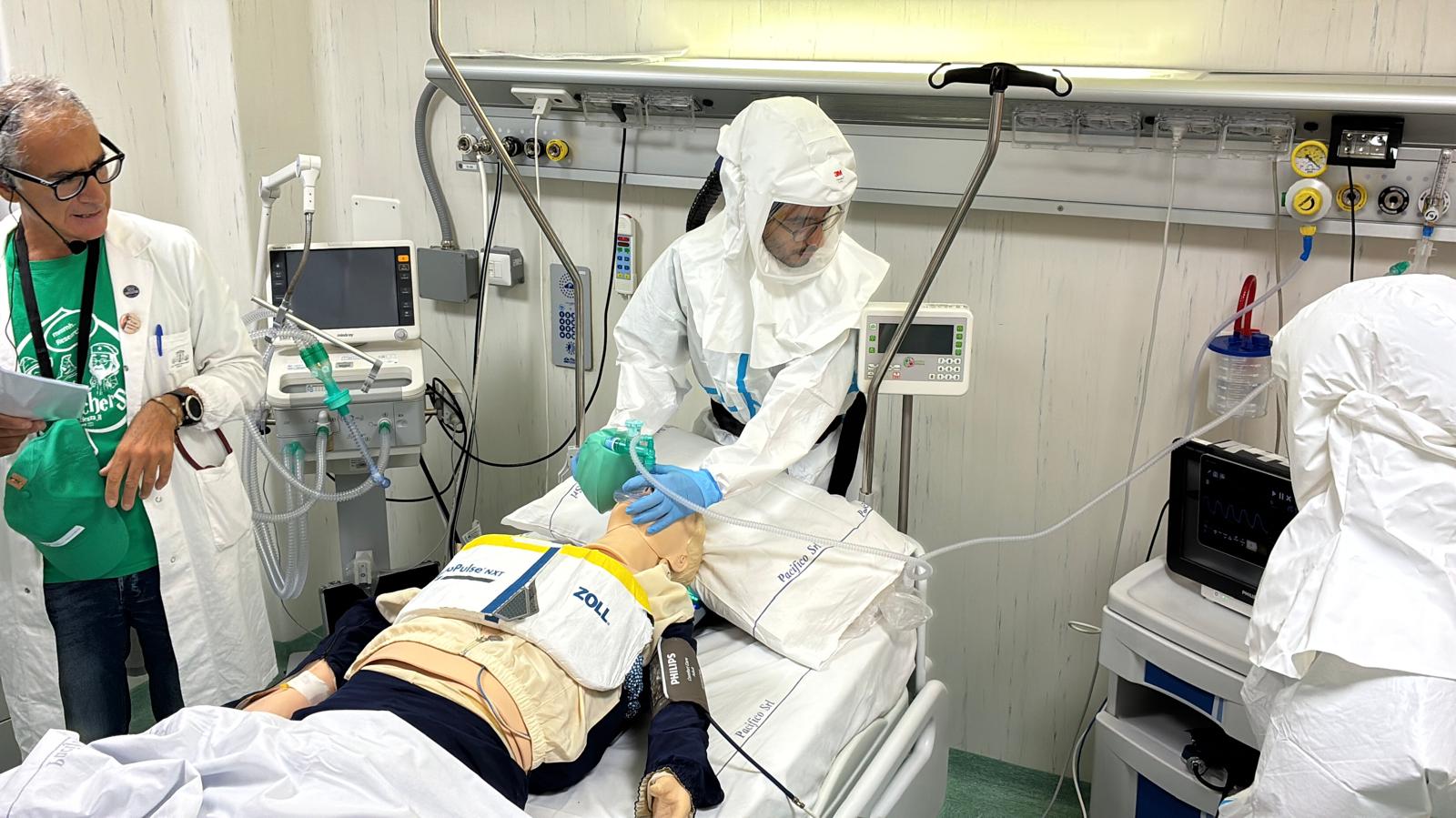
Emergenze infettive, alto isolamento e biocontenimento: Spallanzani e Aeronautica Militare insieme per formare gli operatori sanitari

Malattie venose, Servier lancia Daflon 1000 mg: una sola compressa per semplificare la terapia

Caldo e diabete, il caso Zverev ricorda i rischi dell'estate: "Attenzione ai sensori per la glicemia"
Riflessione di un Tecnico sanitario di radiologia medica