
Tavolo pharma al Mimit: obiettivo rendere l’Italia attrattiva in Europa aspettando la nuova legislazione
27 Marzo 2023
© Riproduzione riservata

Stati Uniti. L’Hhs revoca le autorizzazioni per l’uso in emergenza dei prodotti contro il Covid
Il Dipartimento della Salute e dei Servizi Umani degli Stati Uniti (Hhs) ha annunciato che il segretario Robert F. Kennedy Jr. ha firmato le determinazioni che pongono fine alle dichiarazioni di autorizzazione...

Scienza sotto assedio: perché la fiducia pubblica vacilla tra pandemia, politica e disinformazione
Negli ultimi dieci anni, con un deciso picco nel periodo post-pandemia da Covid-19, si è assistito a un marcato crollo della fiducia della popolazione verso la scienza. Un fenomeno che...

Stati Uniti. Dalla Fda via libera alla prima terapia genica per bambini con anemia falciforme
La Food and Drug Administration (Fda) degli Stati Uniti ha concesso l'approvazione supplementare per Casgevy (exagamglogene autotemcel) per pazienti di età pari o superiore a 2 anni affetti da anemia falciforme con...

Virus respiratori. Si conclude il progetto europeo SeCOV+: rafforzata in Italia la sorveglianza genomica e l’analisi dei dati
Un maggiore raggiungimento degli standard di qualità da parte dei laboratori del network, l’apertura al sequenziamento di nuovi agenti infettivi oltre al Sars-Cov-2 come Rsv e influenza, migliori stime della...
Gli speciali

Sanità digitale per garantire più salute e sostenibilità. Ma servono standard e condivisione
I più letti

Caldo e diabete, il caso Zverev ricorda i rischi dell'estate: "Attenzione ai sensori per la glicemia"

Farmaci. Aifa aggiorna quattro Note: ormone della crescita, infertilità, anticoagulanti e Bpco

Medici di famiglia. Bertolaso (Lombardia): “L'accordo per la loro presenza nelle Case della Comunità è un pannicello caldo”

Farmaci. Con il caldo possono diventare inefficaci o dannosi. I consigli di Fedefarma Verona

Focolaio di Salmonella in Europa. I germogli di erba medica al centro dell'epidemia: 109 casi in 11 Paesi
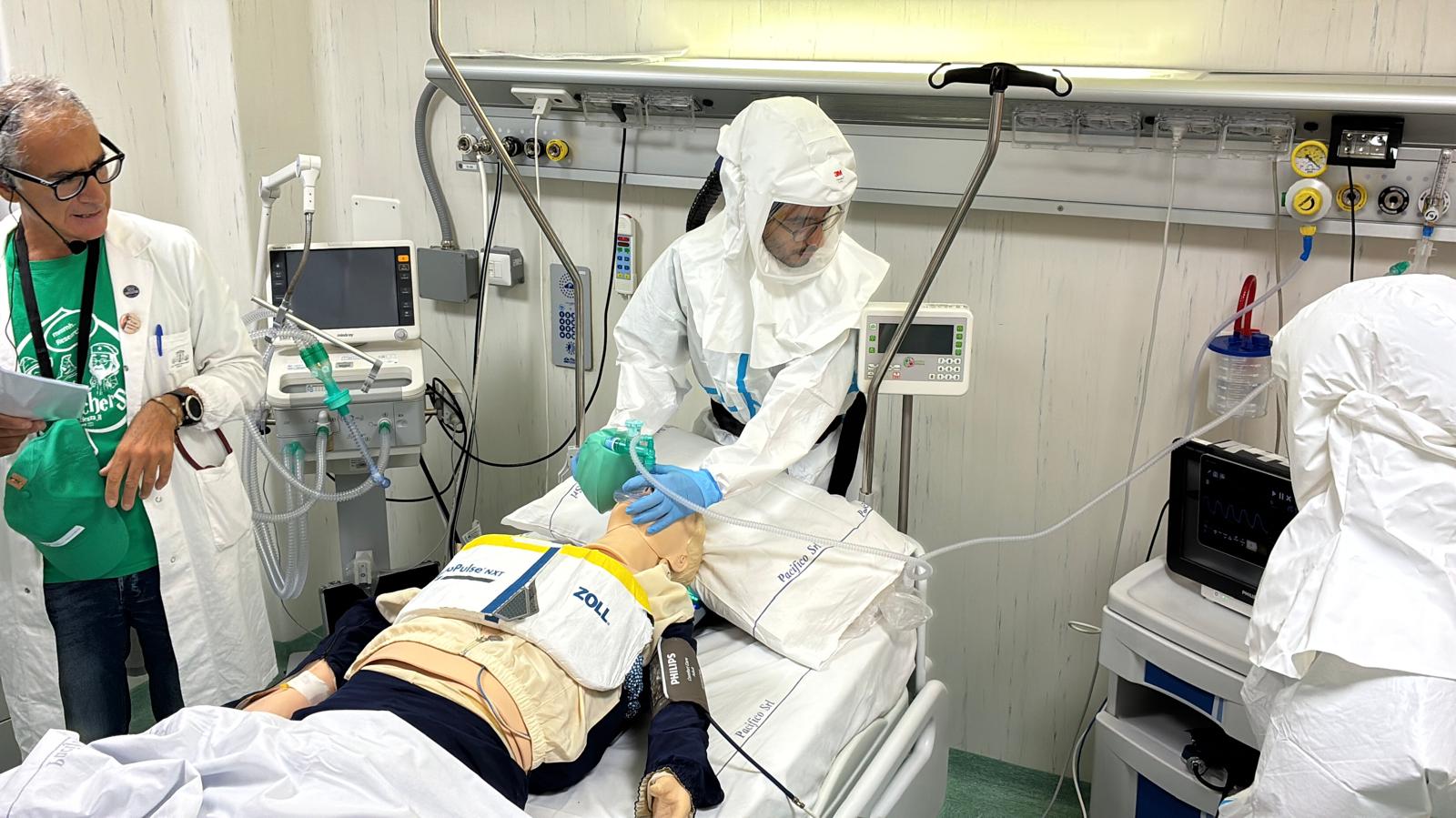
Emergenze infettive, alto isolamento e biocontenimento: Spallanzani e Aeronautica Militare insieme per formare gli operatori sanitari

Malattie venose, Servier lancia Daflon 1000 mg: una sola compressa per semplificare la terapia
Caldo e diabete, il caso Zverev ricorda i rischi dell'estate: "Attenzione ai sensori per la glicemia"

Medico di famiglia. Anatomia di un fallimento

Enpam. Scontro M5S-Giorgetti sulle pensioni dei medici. Castellone: “Investimenti a rischio, c'è un'inchiesta della procura di Milano”