
Da Ema un reflection paper per un uso sistematico dei dati sull’esperienza del paziente nello sviluppo e nella valutazione dei farmaci
29 Settembre 2025
© Riproduzione riservata

Spesa farmaceutica, il tetto non basta. Ma l’Italia deve decidere se governare l’innovazione o continuare a rincorrerla
All’indomani dell’Assemblea di Farmindustria resta una sensazione difficile da scacciare. La politica promette, le imprese chiedono, tutti invocano innovazione, competitività, accesso rapido alle cure, superamento del payback, attrattività del Paese....

Farmaci biosimilari. Dieci anni dalla Legge 232 è allarme sulla corsa al massimo ribasso. Ibg Egualia: “Preservare il modello italiano”
A dieci anni dall’introduzione della Legge 232/2016, l’Italia si conferma tra i Paesi europei con il più elevato utilizzo di farmaci biosimilari e con un modello che ha contribuito ad...

Estate a tavola, dall’Iss le regole per picnic, spiagge e cene all’aperto senza rischi
Pranzi in spiaggia, escursioni in montagna, picnic nei parchi cittadini e cene condivise con amici e familiari. L'estate invita a trascorrere più tempo all'aria aperta e a consumare i pasti...

Prontuario farmaceutico. Federfarma: “Revisione necessaria, ma non penalizzi i cittadini”
“Dopo 30 anni, sicuramente una revisione del Prontuario si rende necessaria. Prima di tutto perché questa revisione è prevista da una legge dello Stato, con obiettivi di ricerca di sostenibilità e...
Gli speciali

Sanità digitale per garantire più salute e sostenibilità. Ma servono standard e condivisione
I più letti
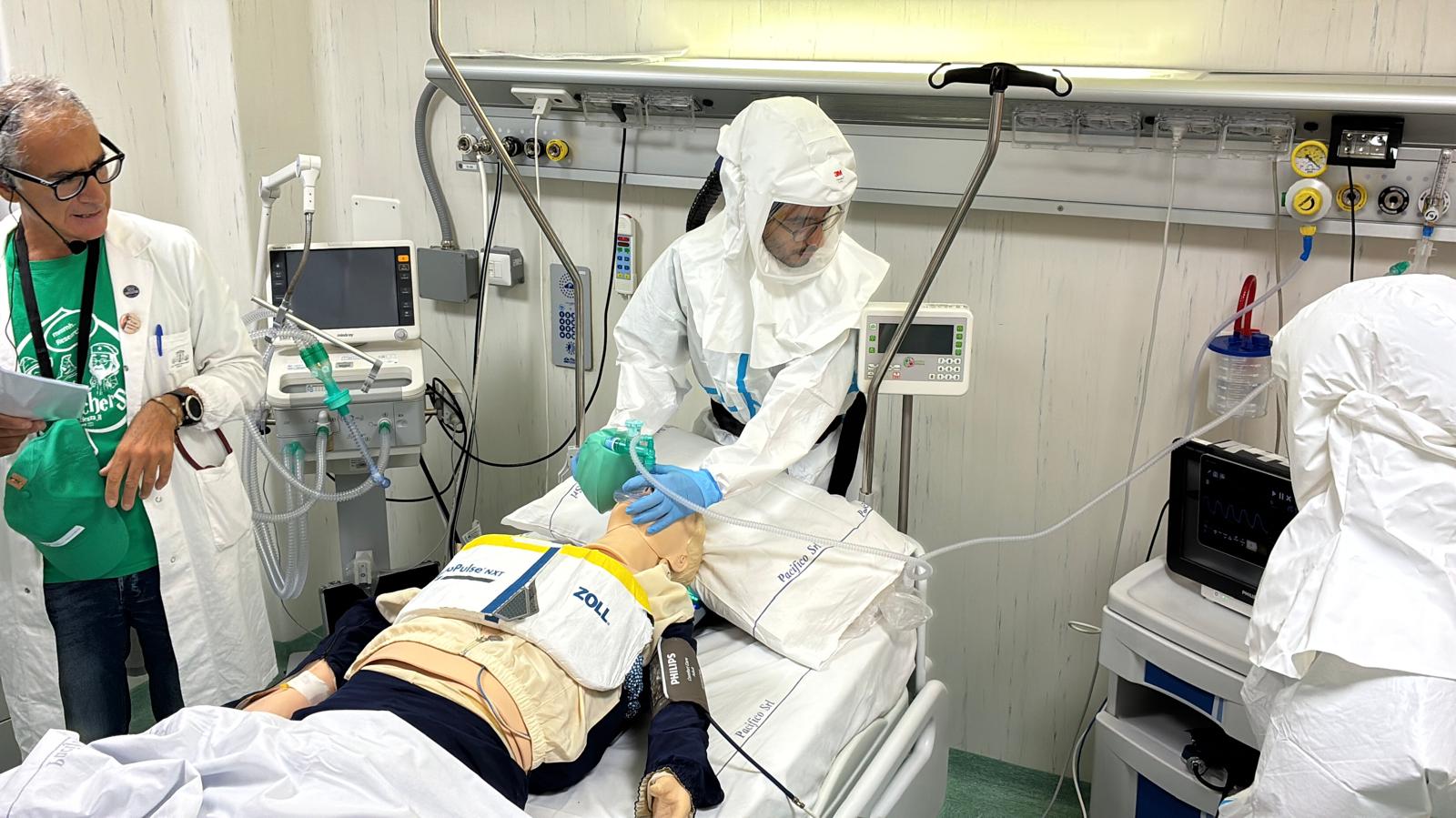
Emergenze infettive, alto isolamento e biocontenimento: Spallanzani e Aeronautica Militare insieme per formare gli operatori sanitari

Malattie venose, Servier lancia Daflon 1000 mg: una sola compressa per semplificare la terapia

Anziani non autosufficienti. Pronto il Piano nazionale 2025-2027: focus su domiciliarità, Pua, Leps e presa in carico integrata

“Il Ssn è governato come una fabbrica di prestazioni. E ogni giorno 10 professionisti lo abbandonano prima della pensione”. L'Anaao Assomed lancia tre proposte per riformare la sanità: “È ora di cambiare modello o crollerà”

Case di Comunità. Per i medici di famiglia un compenso di 38,72 euro l’ora con turni di almeno 3 ore continuative. Arriva la firma sul contratto tra la Sisac e i sindacati Fimmg e Fmt. No di Smi e Snami
Emergenze infettive, alto isolamento e biocontenimento: Spallanzani e Aeronautica Militare insieme per formare gli operatori sanitari
Malattie venose, Servier lancia Daflon 1000 mg: una sola compressa per semplificare la terapia

Rette RSA e Alzheimer: la giurisprudenza svolta. Per la prima volta un Tribunale condanna direttamente anche una Regione

Medico di famiglia. Anatomia di un fallimento

Enpam. Scontro M5S-Giorgetti sulle pensioni dei medici. Castellone: “Investimenti a rischio, c'è un'inchiesta della procura di Milano”