
Farmaci genici. La Fda degli Stati Uniti accelera: “Via i test ridondanti, le terapie salvavita devono arrivare prima ai pazienti”
La nuova bozza di linee guida dell'ente regolatorio americano punta a ridurre i costi e i tempi di sviluppo per le terapie cellulari e geniche destinate a malattie rare e gravi. “Non abbassiamo gli standard, aumentiamo l'efficienza collettiva”.
03 Giugno 2026
© Riproduzione riservata

Aifa. Il Servizio “Cerca un farmaco” si amplia con le informazioni sullo stato di commercializzazione dei medicinali
Il servizio di informazione sui medicinali ad uso umano autorizzati in Italia “AIFA Medicinali”, accessibile anche dalla home del portale AIFA con un clic su “Cerca un farmaco”, si arricchisce...

Estate e arbovirosi, SItI: “Rischio di infezioni è realtà. Costruire modello di prevenzione moderno e sostenibile”
Con una stagione estiva fatta di temperature elevate, ondate di calore frequenti e condizioni favorevoli anche alla proliferazione dei vettori, al centro dell'attenzione tornano le arbovirosi, ovvero le malattie trasmesse...

HIV pediatrico, dall’OMS nuove indicazioni sui dosaggi degli antiretrovirali
L’Organizzazione Mondiale della Sanità ha pubblicato nuove raccomandazioni sui dosaggi dei farmaci antiretrovirali in neonati, lattanti e bambini. L’obiettivo è rafforzare la prevenzione e il trattamento dell’HIV nei pazienti più...

VIVISOL rinnova il sostegno alla Spiaggia dei Valori: al via il progetto “Un mare, mille granelli”
VIVISOL rinnova e amplia il proprio sostegno alla Spiaggia dei Valori di Marina di Ravenna, il progetto dell’associazione"Insieme a te" che rende il mare accessibile anche alle persone con disabilità...
Gli speciali

Sanità digitale per garantire più salute e sostenibilità. Ma servono standard e condivisione
I più letti

Riflessione di un Tecnico sanitario di radiologia medica

Lombardia. Le immagini diagnostiche sul Fascicolo Sanitario Elettronico

Cancro. Oms-Iarc: una persona su 5 svilupperà la malattia. Entro il 2050 attesi 35 mln di nuovi casi l’anno. Quasi il 40% è prevenibile, ma il vero divario è tra ciò che sappiamo e ciò che facciamo

Professionisti sanitari. Via alla raccolta di firme per abolire il vincolo di esclusività: "Facciamo sentire la nostra voce"

Lea 2024: il Nord guida e il Sud insegue. Veneto, Emilia-Romagna e Toscana si confermano al top. Calabria, Sicilia e Bolzano in fondo alla classifica

Benzodiazepine. Stop alla ricetta elettronica: funzionalità sospesa dopo il caos organizzativo
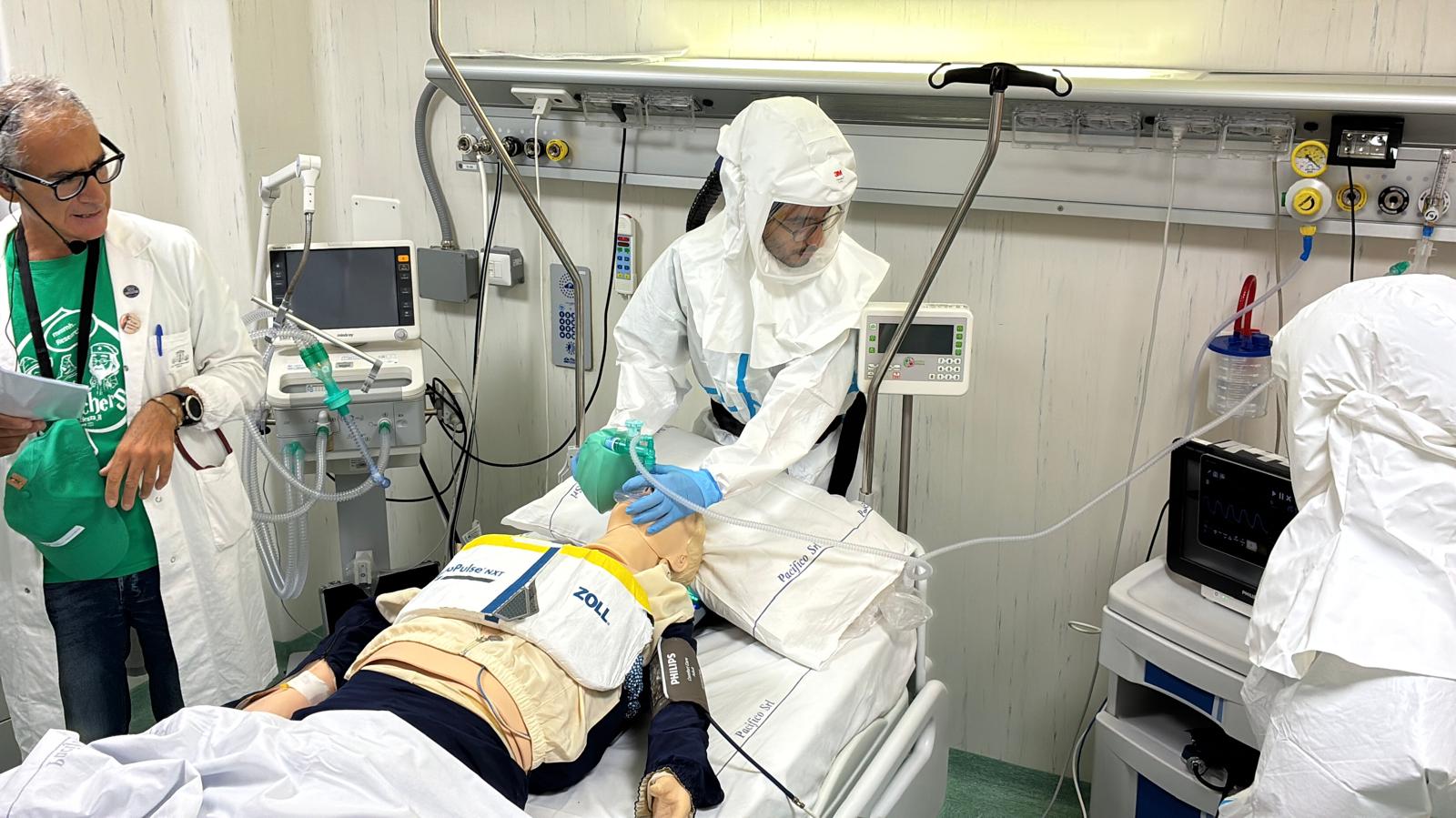
Emergenze infettive, alto isolamento e biocontenimento: Spallanzani e Aeronautica Militare insieme per formare gli operatori sanitari

Malattie venose, Servier lancia Daflon 1000 mg: una sola compressa per semplificare la terapia

Caldo e diabete, il caso Zverev ricorda i rischi dell'estate: "Attenzione ai sensori per la glicemia"
Riflessione di un Tecnico sanitario di radiologia medica