
Da Ema un reflection paper per un uso sistematico dei dati sull’esperienza del paziente nello sviluppo e nella valutazione dei farmaci
29 Settembre 2025
© Riproduzione riservata

Oms Europa: “Il caldo estremo minaccia la salute”. Nuove linee guida per proteggere i cittadini
L'Europa è il continente che si riscalda più rapidamente al mondo, con temperature in aumento a una velocità doppia rispetto alla media globale. Un fenomeno che, secondo l'Organizzazione Mondiale della...

EMA: “Nuova riforma farmaceutica occasione storica per rendere l’Europa più innovativa, competitiva e vicina ai pazienti”
La riforma della legislazione farmaceutica europea rappresenta "la più significativa trasformazione del quadro regolatorio degli ultimi vent'anni" e offre all'Unione europea l'opportunità di costruire un sistema più efficiente, capace di...

Ema e Eismea: intesa per accelerare l’innovazione in sanità
L'Agenzia Europea per i Medicinali (Ema) e l'Agenzia Esecutiva del Consiglio Europeo per l'Innovazione e le PMI (Eismea) hanno rafforzato la loro cooperazione a sostegno dell'innovazione sanitaria nell'Unione Europea. Ieri,...

Estate, SINPIA: “Attenzione ad alterare il ritmo sonno-veglia e all’uso prolungato di strumenti digitali”
Con l'estate e le vacanze, spesso cambiano gli orari delle famiglie. Più occasioni di socialità, tempo all'aperto e attività fisica possono portare ad uno slittamento sistematico dell'addormentamento e, insieme, ad...
Gli speciali

Sanità digitale per garantire più salute e sostenibilità. Ma servono standard e condivisione
I più letti

Riflessione di un Tecnico sanitario di radiologia medica

Lea 2024: il Nord guida e il Sud insegue. Veneto, Emilia-Romagna e Toscana si confermano al top. Calabria, Sicilia e Bolzano in fondo alla classifica

Lombardia. Le immagini diagnostiche sul Fascicolo Sanitario Elettronico

Ebola. Secondo operatore umanitario statunitense contagiato, trasferito in Germania. Tedros: “Proteggere chi è in prima linea”

Medici specialisti, Regioni chiedono di formarne 14.631 nel 2025/2026. Priorità ad anestesia, emergenza-urgenza e medicina interna

Benzodiazepine. Stop alla ricetta elettronica: funzionalità sospesa dopo il caos organizzativo
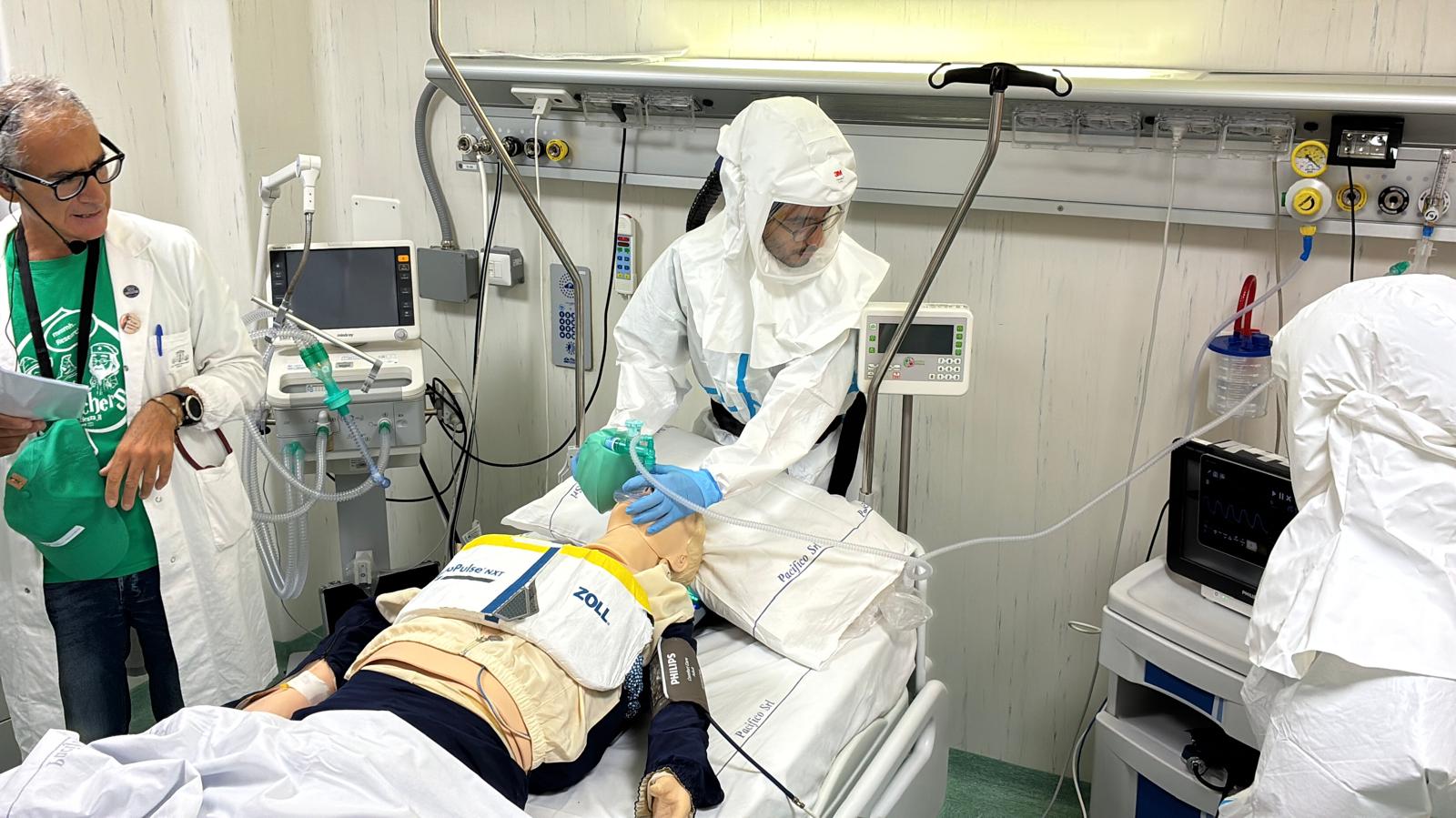
Emergenze infettive, alto isolamento e biocontenimento: Spallanzani e Aeronautica Militare insieme per formare gli operatori sanitari

Malattie venose, Servier lancia Daflon 1000 mg: una sola compressa per semplificare la terapia

Caldo e diabete, il caso Zverev ricorda i rischi dell'estate: "Attenzione ai sensori per la glicemia"
Riflessione di un Tecnico sanitario di radiologia medica